NH3 molecular tight-binding model#
This short notebook walks through a zero-dimensional tight-binding model of an ammonia (\(\text{NH}_3\)) molecule.
What you will learn
Build a
Latticethat hosts four orbitals (one on nitrogen, three on hydrogen).Populate a tight-binding Hamiltonian with onsite terms and uniform hoppings with
TBModel.Visualize the bonds
Diagonalize the Hamiltonian and inspect the molecular levels.
from pythtb import TBModel, Lattice
import numpy as np
import matplotlib.pyplot as plt
Define the molecular lattice#
Even for a 0D system we provide a dummy set of lattice vectors (the identity) so PythTB can keep track of orbital positions.
The three hydrogens are arranged in a trigonal pyramid around the nitrogen.
# Cartesian axes (identity) keep the bookkeeping simple for a 0D molecule
lat_vecs = np.eye(3)
# Reduced coordinates of the four orbitals: one nitrogen at the origin, three hydrogens around it
orb_vecs = np.array(
[
[0, 0, 0], # nitrogen s
[np.sqrt(3) / 3, 0, -1], # hydrogen s
[-np.sqrt(3) / 6, 1 / 2, -1], # hydrogen s
[-np.sqrt(3) / 6, -1 / 2, -1], # hydrogen s
],
dtype=float,
)
# A finite model has no periodic directions:
# we can specify an empty list (default) or None
lat = Lattice(lat_vecs, orb_vecs, periodic_dirs=[])
Build the tight-binding Hamiltonian#
We split the nitrogen onsite energy from the hydrogens with delta and keep all hoppings equal for simplicity.
Tip
Feel free to tweak delta or the hopping strength t to see how the spectrum responds.
model = TBModel(lattice=lat, spinful=False)
eps_N = -5.0 # onsite Nitrogen
eps_H = 0.0 # onsite Hydrogen
t = -2.0 # hopping N–H
# Basis defined in same order as specified orbital vectors
model.set_onsite([eps_N, eps_H, eps_H, eps_H])
# N is site 0; H sites are 1, 2, 3. Only N–H hops.
model.set_hop(t, 0, 1)
model.set_hop(t, 0, 2)
model.set_hop(t, 0, 3)
print(model)
The summary above confirms four orbitals, no periodic directions, and the onsite/hopping configuration we expect.
Let’s visualize the connectivity to verify the symmetry.
model.visualize_3d()
Diagonalize the Hamiltonian#
TBModel.solve_ham() returns the molecular eigenvalues (and eigenvectors if requested).
We display the spectrum and annotate the bonding/antibonding character.
evals, evecs = model.solve_ham(return_eigvecs=True)
fig, ax = plt.subplots()
ax.plot(range(1, len(evals) + 1), evals, "o", color="#1f77b4")
ax.axhline(0.0, linestyle="--", color="0.7", linewidth=1)
ax.set_xticks(range(1, len(evals) + 1))
# ax.set_xticklabels([r"$E_1$", r"$E_2$", r"$E_3$", r"$E_4$"])
ax.set_xlim(0.5, len(evals) + 0.5)
ax.set_ylabel("Energy (arb. units)")
ax.set_title("NH$_3$ molecular energy levels")
plt.show()
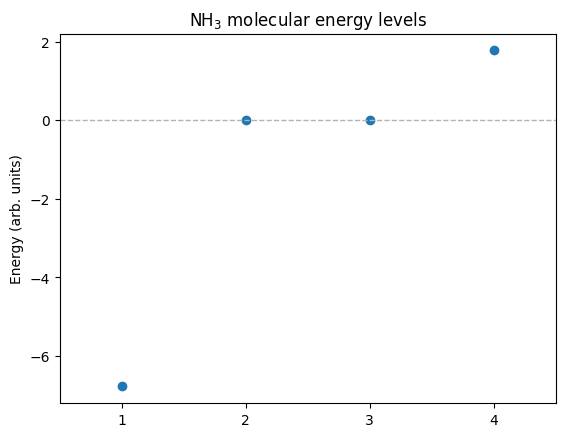
Energy levels (minimal \(\text{NH}_3\) TB model)
With one N orbital and three H orbitals (only N–H hopping \(t\)), the (\(\text{C}_{3v}\)) symmetry yields:
\(\text{A}_1\) bonding (lowest): mixing of N with the symmetric H combination.
\(\text{E}\) doublet (middle, twofold degenerate): non-bonding in this minimal model.
\(\text{A}_1\) antibonding (highest).
Analytic energies:
So the spectrum is ordered: \(\text{A}_1\) (bonding) < \(\text{E}\) (doublet) < \(\text{A}_1\) (antibonding).
E_analytic_plus = (eps_N + eps_H) / 2 + np.sqrt(((eps_N - eps_H) / 2) ** 2 + 3 * t**2)
E_analytic_minus = (eps_N + eps_H) / 2 - np.sqrt(((eps_N - eps_H) / 2) ** 2 + 3 * t**2)
print(f"Analytic energies: {E_analytic_minus}, {eps_H}, {eps_H}, {E_analytic_plus}")
print(f"Computed energies: {evals[0]}, {evals[1]}, {evals[2]}, {evals[3]}")
Analytic energies: -6.772001872658765, 0.0, 0.0, 1.7720018726587652
Computed energies: -6.772001872658766, 0.0, 0.0, 1.7720018726587654
Next steps#
Try
Shift one of the hydrogen onsite energies to mimic distortions or external fields. Notice what happens to the degenerate energy levels.
Tile \(\text{NH}_3\) units into a lattice (
dim_k>0) and plot bands.Reduce the N–H hopping on one bond to see how the spectrum changes.
Make spinful model
spinful=Trueand include spin–orbit terms.